Exploring Catalysis Complexity: A Breakthrough in Atomic Simulations
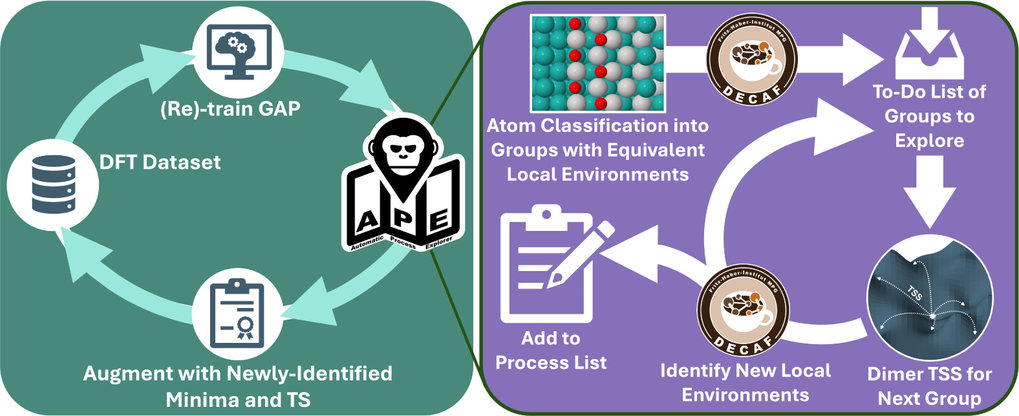
**The Fritz Haber Institute researchers** have taken a giant leap in understanding atomic and molecular processes with the development of the **Automatic Process Explorer (APE)**. Unlike traditional **Kinetic Monte Carlo (kMC) simulations** that rely on static inputs, APE dynamically updates process lists, effectively reducing biases and uncovering overlooked atomic movements. This innovative approach revealed almost 3,000 processes during the oxidation of **Palladium (Pd) surfaces**, which are critical in catalytic converters used to reduce automotive emissions. **APE's success lies in its integration with machine-learned interatomic potentials (MLIPs)**, which predict atomic interactions with high accuracy. By employing fuzzy machine-learning classification, APE identifies distinct atomic environments, enabling a broad exploration of potential atomic movements. This method not only refines traditional simulations but also promotes diversity and efficiency in exploring new atomic structures. The application of APE to Pd surfaces revealed atomic motions and restructuring processes that were previously undetectable. This newfound understanding of Pd oxidation can lead to the development of more efficient catalysts, which play a crucial role in **energy production** and **pollution control**. As a result, APE's insights have the potential to significantly enhance cleaner technologies and promote more sustainable industrial processes.